6ЫъЕФаЁгхЃЈЛЏУћЃЉЃЌгаИіМђЕЅЕФдИЭћЁЊЁЊШУАжАжБГвЛЯТЃЌПЩвЊТњзуетИідИЭћЃЌгаЕуФбЁЃдРДЃЌаЁгхЕФЖЧзгжзеЭЕУЯёЛГдаЦпАЫИідТЕФдаИОЃЌбќзЕЖМБЛМЗбЙЕУБфаЮСЫЃЌИљБОУЛЗЈзіИЉЮдЕФЖЏзїЁЃвђЮЊЫћЛМЩЯСЫИъаЛВЁЃЌвЛжжБШЁАДЩЭоЭоЁБЁЂЁАНЅЖГШЫЁБИќВЛЮЊШЫжЊЃЌШЋЙњЛМепНі300грР§ЁЃаЁгхЕФИИЧзКњаЫЯЃЭћЃЌетбљЕФКБМћВЁФмФЩШыДѓВЁвНБЃЃЌШУКЂзгФмЛюЕУОУвЛЕуЁЃ
ДгаЁЖЧзгДѓШЗеяИъаЛВЁ
дкжиЧьдЦбєЯибпЦКеђЧњЯЊДхЃЌИеТњ6ЫъЕФЭСМвзхФаКЂаЁгхеОдкДАЧАПДзХЦфЫћаЁЛяАщУЧцвЯЗЭцЫЃЃЌблРяГфТњСЫЯлФНКЭЦкД§ЁЃЁАЕШФуЖЧзгБфаЁСЫЃЌАжАжОЭДјФуГіШЅЗХЗчѓнЁЃЁБвЛХдЕФИИЧзКњаЫЫЕЭъетОфЛАЃЌдчвбРсблЦХцЖЁЃвђЮЊетбљЕФГаХЕЃЌЫћвбОаэЯТЮоЪ§ДЮЃЌЪВУДЪБКђФмЪЕЯжЃЌЫћаФРявВУЛЕзЁЃ
КњаЫИцЫпМЧепЃЌаЁгхДгаЁЖЧзгОЭБШБ№ЕФаЁКЂДѓЃЌЕЋЦ№ГѕМвШЫВЂУЛдквтЁЃдкаЁгх4ЫъЪБЃЌМвШЫЗЂЯжЫћЖЧзгдНРДдНДѓЛЙгаЕугВЃЌЫФжЋШДКмЪнЃЌЛЙОГЃСїБЧбЊЃЌБуДјЫћЕНЕБЕивНдКМьВщЃЌНсЙћЗЂЯжаЁгхЖЧзгБфДѓЪЧвђЦЂдржзеЭЫљжТЃЌЕЋОпЬхдвђВЂВЛУїШЗЁЃЁАЪдЙ§КмЖрАьЗЈЃЌГдСЫКмЖрвЉЃЌЛЙЪЧУЛгаЦ№ЩЋЁЃЁБКњаЫЫЕЃЌзюКѓЫћОіЖЈАбКЂзгДјЕНЙужнЪаИОХЎЖљЭЏвНдКМьВщЃЌаЁгхзюжеБЛШЗеяЮЊЁАИъаЛВЁЁБЁЃ
вЛЬьвЊЩЯЖўЪЎЖрДЮВоЫљ
6ЫъЕФаЁгхЬхжиВЛЕН40НяЃЌГ§СЫвђжзДѓЕФЦЂдрЖјИпИпТЁЦ№ЕФЖЧзгЯёЦпАЫИідТЕФдаИОЃЌЩэЬхЦфЫћВПЮЛЖМЪЎЗжЪнШѕЁЃвНЩњНщЩмЃЌИъаЛВЁЪЧвђЬхФкЯШЬьШБЗІвЛжжУИЃЌНсЙћЕМжТЖрЯЕЭГЕФжЌжЪРлЛ§ЃЌдьГЩЖржжЦїЙйЪмРлЗЂВЁЃЛИЮЦЂжзДѓЁЂИЮЙІФмвьГЃЁЂбЊЯИАћМѕЩйЃЌЖљЭЏЩњГЄЗЂг§жЭЛКЕШЪЧГЃМћжЂзДЁЃ
гЩгкжзДѓЕФЦЂдрбЙЦШАђызЃЌаЁгхвЛЬьвЊЩЯ20ЖрДЮВоЫљЁЃвђЮЊбЊаЁАхМЋЕЭЃЌЦЂдржзДѓгжЬиБ№бЯжиЃЌШЮКЮЕФХізВзїгУЃЌЖМгаПЩФмдьГЩаЁгхДѓГібЊКЭЦЂдрЦЦСбЁЃ
КњаЫИцЫпМЧепЃЌаЁгхЫфШЛадИёФкЯђЃЌШДЗЧГЃЯВЛЖЩЯбЇЃЌгШЦфЯВЛЖжщаФЫуЁЃЖјдквЛМвШЫОгзЁЕФЦЦОЩИѓТЅЧНЩЯЃЌЮЈвЛЕФзАЪЮОЭЪЧЫћКЭЖСаЁбЇЕФИчИчЕУЕНЕФаэЖрНБзДЁЃ
вЉЗбУПИідТОЭвЊ7ЭђдЊ
вНЩњИцЫпКњаЫЃЌетВЁЪЧгавЉПЩвджЮСЦЕФЃЌОЭЪЧАбвЛжжЬцДњУИзЂЩфНјаЁгхЬхФкАяжњаТГТДњаЛЁЃетИіЯћЯЂШУКњаЫЫЦКѕПДЕНСЫЪяЙтЁЃЕЋвНЩњИцЫпЫћЃЌетжжвЉЗЧГЃАКЙѓЃЌЯёаЁгхвЛИідТвЊгУШ§ЦПвЉЃЌЛЈЗбОЭвЊНќ7ЭђдЊЁЃЖјЫцзХФъСфдіДѓЃЌКЂзгЬхжиЕФдіМгЃЌетжжвЉЕФгУСПЛЙвЊМгДѓЃЌГЩФъШЫвЛФъгУвЉЛЈЗбПЩФмвЊ200ЭђдЊЁЃЮЊСЫељШЁИјКЂзгжЮСЦЕФЛњЛсЃЌКњаЫжЛгаЕНДІНшЧЎЁЃШчНёЃЌ80КѓЕФКњаЫСГЩЯдчвбОжхЮЦДдЩњЁЃ
КњаЫЬиБ№ЯЃЭћжиЧьФмНЋИъаЛВЁЕФжЮСЦЗбвВФЩШывНБЃКЭУёеўОШжњЗЖЮЇФкЃЌШУаЁгхПЩвдОЁдчНгЪмжЮСЦЁЃЮЊСЫдВУЮЃЌетЮЛФъЧсЕФИИЧзПЊЪМЫФДІБМзпЃЌДгЯчеўИЎЕНЯиеўИЎЃЌЩѕжСЧѓжњгкУНЬхЁЃ
КБМћВЁгаЭћНјДѓВЁвНБЃ
ЖдДЫЃЌжиЧьЪаШЫЩчОжвНБЃДІЯрЙиИКд№ШЫБэЪОЃЌКБМћВЁга7000ЖржжЃЌЖјжЛга1%ЕФКБМћВЁгаЬиаЇвЉжЮСЦЁЃЖдетаЉгагааЇжЮСЦвЉЮяЕФКБМћВЁЃЌвНБЃВПУХвВдкЛ§МЋКЭвЉЦѓЬИХаЃЌЯЃЭћФмАбвЉМлНЕЯТРДЃЛСэвЛЗНУцЃЌдђдкОЁСІЭъЩЦКБМћВЁЕФвНвЉЗбБЃеЯЛњжЦЃЌБШШчНЋКБМћВЁФЩШыДѓВЁвНБЃЕШЃЌЁЃ
ИУИКд№ШЫЛЙБэЪОЃЌФПЧАИъаЛВЁЩаЮДНјШыДѓВЁвНБЃФПТМЃЌЛМепМвЪєПЩЯђЕБЕиЩчБЃОжЬсГіЩъЧыЃЌЩчБЃВПУХКЫЪЕКѓЬсНЛЩЯМЖОЙ§баОПКѓдйжЦЖЈвНСЦБЃеЯЛњжЦЃЌКБМћВЁСаШыДѓВЁвНБЃВЂВЛЪЧВЛПЩФмЕФЁЃ
зЈМвНтЖС
ИъаЛВЁЗЂВЁТЪЖўЪЎЭђЗжжЎвЛ
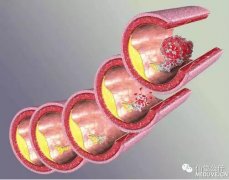
ЁАИъаЛВЁЪЧКБМћВЁжаЕФКБМћВЁЁЃЁБЮїФЯвНдКВњЧАеяЖЯжааФвХДЋбЇзЈМввІКъНЬЪкИцЫпМЧепЃЌЫќЪЧвЛжжгЩЛљвђЭЛБфв§ЗЂЕФвХДЋВЁЃЌЪЧвдЕквЛИіЗЂЯжИУВЁЕФвНЩњУћзжУќУћЕФЁЃ
ИУВЁЮЊЪВУДШчДЫКБМћЃПЁАИъаЛВЁЕФЗЂВЁТЪЮЊЖўЪЎЭђЗжжЎвЛЁЃЛМепЗЂВЁЕФдвђЃЌЪЧвђЮЊзїЮЊШБЯнЛљвђаЏДјепЕФИИФИе§КУХіЕНСЫвЛЦ№ЃЌвХДЋЕНКЂзгЩэЩЯЗЂВЁЕФПЩФмадОЭга25%ЁЃЁБвІКъНЬЪкНтЪЭЫЕЃЌаЁгхЕФИИФИзїЮЊвўадаЏДјепЃЌга1/4ЕФМИТЪЩњЯТЛМВЁЕФКЂзгЃЌЖјаЁгхе§ЪЧетВЛавЕФ1/4ЁЃФПЧАШЋжаЙњвбжЊШЗеяЕФИъаЛВЁЛМепжЛгадМ300грУћЃЌЖјдкДЫжЎЧАЃЌжиЧьгаЛЙУЛгаМЧдиЕФИъаЛВЁЛМепЁЃ
КБМћВЁЕФвЉЮЊКЮетУДЙѓ
вІКъНЬЪкНщЩмЃЌФПЧАШЋЪРНчБЛШЯжЊЕФМВВЁга14000ЖржжЃЌКБМћВЁга7000ЖржжЃЌЖјжЛга1%ЕФКБМћВЁгаЬиаЇвЉжЮСЦЃЌФПЧАжЮСЦКБМћВЁЕФвЉЮяБЛГЦЮЊЁАЙТЖљвЉЁБЃЌДѓЖрВЩгУЩњЮяММЪѕбаЗЂКЭЩњВњЁЃБШШчжЮСЦИъаЛВЁЕФвЉЪЧДгЬЅХЬжаЬсШЁЕФЃЌЭтМгЪЧеыЖдКБМћВЁЃЌгУвЉЪаГЁЗЧГЃаЁЃЌЫљвдМлИёЗЧГЃАКЙѓЁЃвІКъНЬЪкЛЙЛ§МЋКєгѕЩчЛсИјетаЉКБМћВЁЛМепИќЖрЕФЙиАЎЃЌеўИЎВПУХФмЬсЙЉеўВпЗіГжЁЃ